Moderne Technik wie die Magnet-Resonanz- Tomographie (MRT) macht es möglich, die Struktur, Funktion und den Stoffwechsel des Gehirns zu erforschen. Im Bereich der neurodegenerativen Erkrankungen, denen eine Jahre oder sogar Jahrzehnte andauernde Phase ohne beobachtbare klinische Symptome vorangehen kann, eröffnet die MRT-Bildgebung vielfältige Möglichkeiten, das Prodromalstadium (Phase, in der uncharakteristische Frühsymptome auftreten) und die frühen manifesten Stadien neurodegenerativer Erkrankungen näher zu erforschen. Denn zum Zeitpunkt der klinischen Diagnose zeigen sich bereits deutliche Veränderungen in bestimmten Bereichen des Gehirns. Viele innovative Therapien bei neurodegenerativen Erkrankungen könnten bisher daran gescheitert sein, dass die Behandlung zu spät begonnen hat. Darum ist die Identifizierung von unter anderem bildgebenden Markern, die Vorboten von neurodegenerativen Erkrankungen sein können, unverzichtbar.
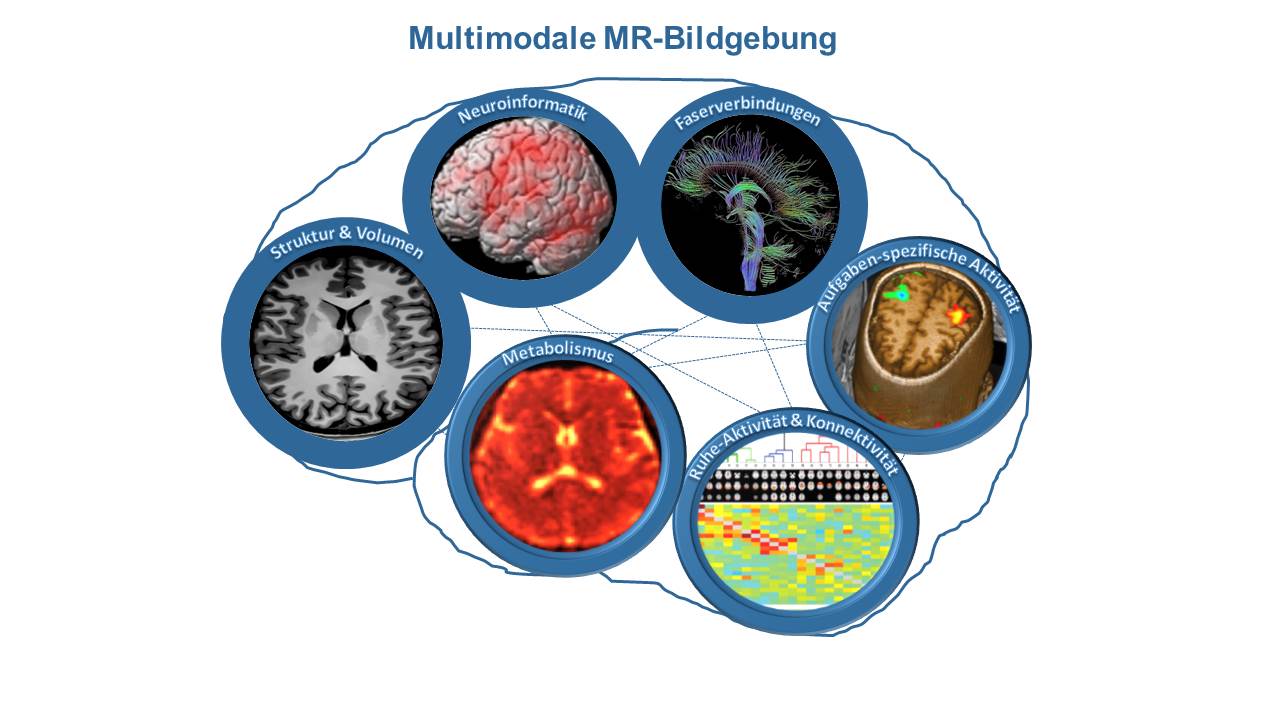
Die Forschungsgruppe um Univ.-Prof. Dr. med. habil. Kathrin Reetz, Oberärztin der Klinik für Neurologie an der Uniklinik RWTH Aachen, beschäftigt sich daher mit der Identifizierung von krankheitsspezifischen Bildgebungsmarkern für neurodegenerative Erkrankungen durch innovative bildgebende Verfahren und deren Bewertung im Kontext klinischer und genetischer Parameter (Abbildung). Ein besonderer Schwerpunkt liegt hierbei auf den seltenen, meist genetisch bedingten, neurodegenerativen Erkrankungen. Ziel ist es, ein besseres pathophysiologisches Verständnis neurodegenerativer Erkrankungen zu gewinnen, um eine verbesserte Vorhersage individueller Erkrankungsrisiken und -verläufe zu ermöglichen. Diese Arbeit erfolgt im Rahmen der Jülich-Aachen Research Allianz (JARA-BRAIN) in enger Zusammenarbeit mit dem Forschungszentrum Jülich. Gerade bei erblichen Ataxien mit dem Leitsymptom der ataktischen Gangstörung, der häufigsten autosomal-dominanten spinocerebellären Ataxien (SCA), als auch bei der autosomal-rezessiv vererbten Friedreich Ataxie, kommt der MRT-Bildgebung und ihrer Vielfalt eine besondere Bedeutung zu.
Erbliche Ataxien
Die autosomal-dominant vererbten Ataxien werden nach genetischer Nomenklatur als spinocerebelläre Ataxien (SCA) bezeichnet. Die SCA manifestieren sich üblicherweise im mittleren Erwachsenenalter mit allmählich fortschreitender Störung der Bewegungskoordination (cerebelläre Ataxie), die entweder allein oder in Kombination mit anderen neurologischen Symptomen wie Parkinson-Syndrom, Dystonie, Spastik, Schluckstörung, Sakkadenverlangsamung, Ophthalmoplegie, Neuropathie, autonomer Dysfunktion oder kognitiver Störung auftritt.
Große multizentrische Registerstudien für seltene Erkrankungen ermöglichen laborchemische und klinische Verlaufsuntersuchungen. Weitere Messmöglichkeiten kommen aus dem Bereich der Neurobildgebung.
Im Rahmen der Ataxia-Study-Group konnten in den Projekten für manifeste spinocerebelläre Ataxien „European Spinocerebellar Ataxia Registry (EuroSCA)“ und für Genträger im prämanifesten Stadium „Prospective Study of Individuals at Risk for Spinocerebellar Ataxie (RISCA)“ klinische und bildgebende MRT-Daten erhoben werden. Im manifesten Stadium der häufigsten SCAs (1, 2, 3, und 6) zeigten sich mittels volumetrischer Auswertung der strukturellen MRT-Daten Genotyp-abhängige Hirnvolumenminderungen hauptsächlich im Hirnstamm und Kleinhir (7). In der 2-Jahres-MRT-Verlaufsstudie hat die Forschungsgruppe Genotyp-abhängige Veränderungen des Hirnvolumens nachgewiesen, die ähnlich gut hinsichtlich ihrer Effektstärke als der beste klinische Marker zur Messung der Ataxie waren (3).
Im präsymptomatischen Stadium der häufigsten SCA-Typen hat das Team um Prof. Reetz in der europäischen multizentrischen RISCA-Studie mit der strukturellen MRT bei den SCA Typen 1 und 2 ebenfalls erste leichtgradige Veränderungen im Hirnstamm und Kleinhirn nachgewiesen (2). Diese Veränderungen des Hirnvolumens konnte für die SCA2 mit einer deutlich größeren Personengruppe aus Kuba (aufgrund eines Gründereffektes kommt diese Erkrankung dort außerordentlich häufi g vor) nun nochmals bestätigt werden(6).
„Mit verschiedenen Bildgebungsverfahren wie der Kernspintomografie haben wir die Möglichkeit, bereits im präsymptomatischen Stadium, sprich vor Ausbruch der Krankheit, erste Veränderungen im Gehirn vorzeitig nachzuweisen. Wir müssen diese ersten Veränderungen identifizieren und besser verstehen, um zukünftige Therapien so früh wie möglich durchführen zu können, bevor es zu irreversiblen Schäden kommt“, so Prof. Reetz.
Die Friedreich Ataxie gehört zu den seltenen autosomal-rezessiv vererbten Erkrankungen. Bei diesem Vererbungsmodus sind beide Eltern genetische Anlageträger, erkranken aber im Gegensatz zu ihren Kindern nicht selbst. Klinisch führt sie meist mit Beginn um die Pubertät unter anderem zu unkoordinierten Bewegungsabläufen, Haltungs- und Gangstörungen, Gleichgewichts- und Sehstörungen, aber auch zu Herzmuskelerkrankungen, Diabetes und Skoliose.
Im Rahmen des europäischen Forschungsregisters „European Friedreich‘s Ataxia Consortium for Translational Studies“ unter Leitung von Prof. Schulz, Direktor der Klinik für Neurologie an der Uniklinik RWTH Aachen, wird der natürliche Verlauf der Erkrankung erforscht, um Merkmale der Friedreich Ataxie besser zu verstehen und zukünftige klinische Studien zu etablieren (4;5).
Da bei der Friedreich Ataxie ebenfalls das Rückenmark betroffen ist, wird mithilfe der Kernspintomographie untersucht, inwiefern es auch zu Volumenminderungen des Rückenmarks neben den zerebralen Veränderungen (das Gehirn betreffend) bei der Erkrankung kommt.
„Zusätzlich werden auch funktionelle MRT-Daten erhoben, die Zusammenhänge typischer Symptome wie Motorik, Kognition und psychische Veränderungen untersuchen, wie kürzlich eine Studie zur Kognition bei der Friedreich Ataxie von Dr. Imis Dogan aus unserer Arbeitsgruppe zeigte“ (1), ergänzt die Oberärztin. Doch nicht nur Struktur und Funktion können mit der Kernspintomographie gemessen werden, sondern auch Stoffwechselveränderungen.
Mit der Förderung der Nachwuchsgruppe von Prof. Reetz durch das Bundesministerium für Bildung und Forschung (BMBF) werden darüber hinaus innovative Bildgebungstechniken wie Natrium-MRT und Phosphor-Magnetresonanzspektroskopie gemeinsam mit Dr. Sandro Romanzetti entwickelt, optimiert und eingesetzt, um Stoffwechselveränderungen in besonders frühen Stadien der Erkrankungen zu untersuchen. Erste Untersuchungen mit der Natrium-MRT bei der Friedreich Ataxie zeigen Veränderungen der Natrium-Konzentration im Kleinhirn und Hirnstamm, die spannenderweise auch mit klinischen Parametern der Erkrankung assoziiert scheinen.
Um diese metabolischen Fehlfunktionen, insbesondere in frühen Erkrankungsstadien, besser zu verstehen, wird ein multimodales Bildgebungssetting (Abbildung) angewandt. Hierbei werden neben klinischen und genetischen Parametern, Struktur, Funktion und Metabolismus des zentralen Nervensystems gemessen, um maximal vielfältige Informationen zu gewinnen und den besten Marker oder auch kombinierten Marker zu identifizieren.
„Mit unserer Forschungsarbeit möchten wir zu einem besseren Verständnis der Erkrankungen beitragen. Und natürlich ist es das Ziel, einen ‚bildgebenden Marker‘ für neurodegenerative Erkrankungen zu entwickeln, der uns hilft, den Verlauf und Therapieeffekte auch in frühen Phasen der Erkrankungen zu messen. Insbesondere für die seltenen Erkrankungen ist eine überregionale und länderübergreifende Zusammenarbeit der spezialisierten Zentren extrem wichtig. Nur so werden wir ausreichend Daten und Informationen gewinnen und langfristig zur Optimierung der Therapie und Versorgung von Patienten und Angehörigen beitragen“, macht Prof. Reetz deutlich. ![]()
Referenzen:
1Dogan I, Tinnemann E, Romanzetti S, Mirzazade S, Costa AS, Werner CJ, et al. Cognition in Friedreich‘s ataxia: a behavioral and multimodal imaging study. Ann Clin Transl Neurol 2016; 3(8):572-87.
2Jacobi H, Reetz K, du Montcel ST, Bauer P, Mariotti C, Nanetti L, et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. The Lancet Neurology 2013; 12(7): 650-8.
3Reetz K, Costa AS, Mirzazade S, Lehmann A, Juzek A, Rakowicz M, et al. Genotype-specific patterns of atrophy progression are more sensitive than clinical decline in SCA1, SCA3 and SCA6. Brain : a journal of neurology 2013; 136(Pt 3): 905-17.
4Reetz K, Dogan I, Costa AS, Dafotakis M, Fedosov K, Giunti P, et al. Biological and clinical characteristics of the European Friedreich‘s Ataxia Consortium for Translational Studies (EFACTS) cohort: a cross-sectional analysis of baseline data. The Lancet Neurology 2015; 14(2): 174-82.
5Reetz K, Dogan I, Hilgers RD, Giunti P, Mariotti C, Durr A, et al. Progression characteristics of the European Friedreich‘s Ataxia Consortium for Translational Studies (EFACTS): a 2 year cohort study. The Lancet Neurology 2016; 15(13): 1346-54.
6Reetz K, Rodriguez-Labrada R, Dogan I, Mirzazade S, Romanzetti S, Schulz JB, et al. Brain atrophy measures in preclinical and manifest spinocerebellar ataxia type 2. Annals of Clinical and Translational Neurology in press.
7Schulz JB, Borkert J, Wolf S, Schmitz-Hubsch T, Rakowicz M, Mariotti C, et al. Visualization, quantification and correlation of brain atrophy with clinical symptoms in spinocerebellar ataxia types 1, 3 and 6. NeuroImage 2010; 49(1): 158-68.