Wenn sich Forscher mit einem neuen Krankheitsbild befassen, arbeiten sie manchmal wie Kriminalisten. Zuerst analysieren sie genau den biologischen Mechanismus hinter der Erkrankung, dann suchen sie nach Substanzen, um diesen Mechanismus zu beeinflussen. Wie kompliziert und langwierig muss man sich so eine Medikamentenentwicklung vorstellen? An der Uniklinik RWTH Aachen steht den Forschern dafür das Center for Translational & Clinical Research (CTC-A) zur Seite.

Neun von zehn Bundesbürgern unterschätzen die Dauer und 79 Prozent die Forschungs- und Entwicklungskosten für ein neues Medikament. Tatsächlich vergehen durchschnittlich über 13 Jahre von den ersten Tests im Labor bis zur Zulassung. Pharmaunternehmen investieren im Schnitt rund eine Milliarde Euro dafür. Der Weg zu einem neuen Medikament, das man beim Arzt verschrieben bekommt oder in der Apotheke kaufen kann, ist lang. Er beginnt nicht mit dem Prozess der Marktzulassung, sondern bereits Jahre zuvor bei der Suche, dem Erforschen, dem Testen und Auswerten von Ergebnissen aus den unterschiedlichen Phasen einer Arzneimittelentwicklung. Dabei sind Wirkstoffe wie die sprichwörtliche Nadel im Heuhaufen: Von rund 10.000 bis 15.000 in der Grundlagenforschung und Präklinik (Phase, in der Studien noch nicht am Menschen, sondern im Zell- oder Tiermodell durchgeführt werden) untersuchten Substanzen erhalten am Ende dieses langen Weges nur ein bis zwei Kandidaten die Zulassung der Arzneimittelbehörden.
Zentraler Bestandteil jedes Medikaments ist sein Wirkstoff, also ein Stoff, der im Körper eine heilende oder lindernde Wirkung erzielt. In vielen Fällen beginnt die Suche deshalb mit einer genauen Analyse der Krankheit. An welcher Stelle können Substanzen eingreifen und den Prozess zum Wohl der Betroffenen beeinflussen? „Oft sind es Moleküle wie Enzyme oder Rezeptoren, die als Ziel von Stoffen infrage kommen. Dann erfolgt die Suche nach geeigneten Wirkstoffkandidaten: über Screenings von Substanzbibliotheken, aber auch über die gezielte Synthese von Substanzen, die an das Ziel andocken können. Manchmal liegt es nahe, was ein geeigneter Wirkstoff zur Behandlung einer Krankheit sein könnte: Etwa dann, wenn Patienten deshalb krank sind, weil ihnen eine bestimmte Substanz teilweise oder ganz fehlt“, erklärt Dr. med. Susanne Isfort, Koordinierende Geschäftsführerin des CTC-A.
Vorklinische Entwicklung: Wirkstoffe im Auswahlverfahren
Sicherheit ist immer die zentrale Maxime in der Medikamentenentwicklung. Ehe eine Substanz mit Menschen erprobt werden kann, muss sie ein straffes Prüfprogramm bestehen: die vorklinische Entwicklung. Dazu gehören insbesondere Tests auf schädliche Wirkungen: Toxikologen untersuchen dabei, ob und ab welcher Konzentration die Substanz möglicherweise giftig ist, ob sie Embryonen schädigt, Krebs auslöst oder Veränderungen des Erbguts hervorruft. Manches davon kann im Labor oder mit Zellkulturen untersucht werden, anderes jedoch lässt sich nur an Organismen studieren. Deshalb sind bestimmte Versuche mit mindestens zwei Tierarten gesetzlich vorgeschrieben. Nur Stoffe, die sich hier bewähren, kommen für eine Erprobung mit Menschen überhaupt in Betracht – das dauert eine ganze Weile. Bis zum Abschluss der präklinischen Tests sind üblicherweise schon mehr als fünf Jahre vergangen. Ist die Präklinik erfolgreich, kann es mit der Entwicklung weitergehen.
Klinische Entwicklung: Erprobung mit Menschen bis hin zur Zulassung
In einer ersten klinischen Studie, in der Regel mit wenigen gesunden Erwachsenen, wird zunächst die Verträglichkeit und die Sicherheit getestet. Ausnahmen bilden hier Medikamente gegen Krebs oder ähnlich schwere Erkrankungen; diese müssen direkt bei Patienten getestet werden. Wie verhält sich der Stoff im Körper eines Menschen? Welche Dosis kann ohne Nebenwirkungen verabreicht werden und ist das Medikament sicher? Für diese Studien muss der Wirkstoff zuvor unter speziellen Bedingungen hergestellt werden. Dr. Isfort: „In der Regel nehmen 20 bis 80 Probanden an einer solchen Studie teil. Sind Verträglichkeit und Sicherheit geprüft, geht es in den Phasen II a und II b darum, die Wirksamkeit und die Dosierung genauer zu untersuchen. Dafür wird zunächst die Darreichungsform entwickelt. Bei Phase II a-Studien prüfen die Experten das Therapiekonzept (Anmerkung der Redaktion: Proof of Concept), in Phase II b-Studien geht es ihnen darum, die richtige Dosis zu finden. Phase II-Studien schließen dann in der Regel schon 50 bis 200 erwachsene Erkrankte ein.“ Die Teilnahme ist freiwillig. Jeder Patient wird vorab umfangreich aufgeklärt und muss einer Teilnahme explizit zustimmen. Zudem müssen alle klinischen Studien vor Beginn jeweils von der zuständigen nationalen Behörde und einer Ethik-Kommission genehmigt werden. In der letzten Phase vor einer möglichen Zulassung als Medikament erproben Ärzte das Arzneimittel schließlich an einer größeren Anzahl an Patienten. Die Summe ist abhängig vom Krankheitsbild und kann je nach Krankheit mehrere Tausend Patienten betragen. So kann man sehen, ob sich Wirksamkeit und Unbedenklichkeit bei vielen unterschiedlichen Patienten bestätigen lassen. Dabei werden auch Wechselwirkungen mit anderen Medikamenten erprobt. Manche Phase II-, aber vor allem Phase III-Studien sind typischerweise kontrollierte Studien: Ein Teil der Patienten erhält das neue Mittel, eine andere Gruppe das bisherige Standardpräparat. Auf diese Weise lässt sich der Effekt genau nachweisen.
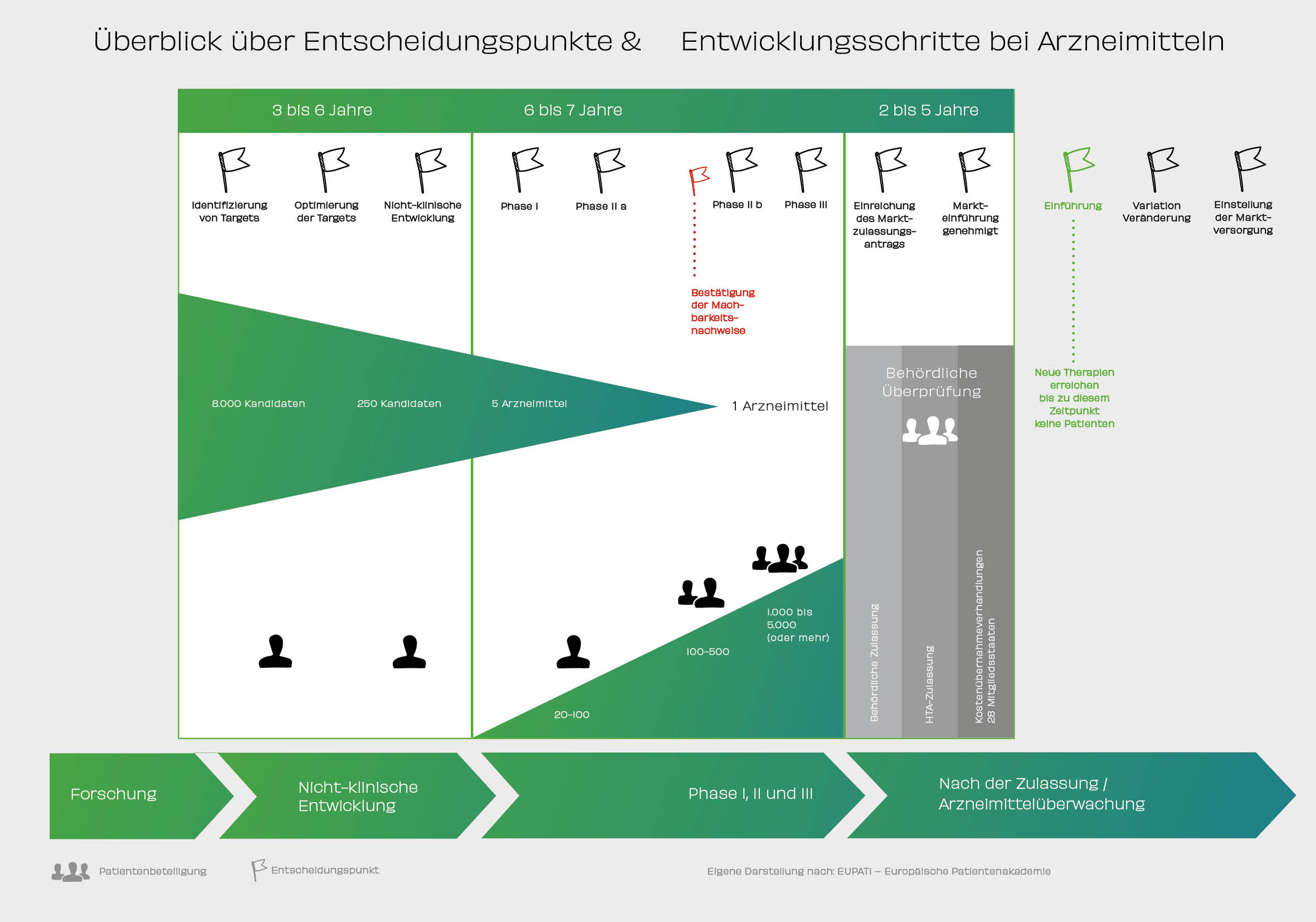
Überblick über Entscheidungspunkte & Entwicklungsschritte bei Arzneitmitteln
Endgültige Zulassung von Medikamenten
Waren alle Studien und Tests erfolgreich, kann der Hersteller bei den Behörden die Zulassung beantragen. Für Länder der Europäischen Union tut er dies bei der europäischen Arzneimittelagentur EMA (European Medicines Agency). In einigen seltenen Fällen kann er den Antrag auch bei einer nationalen Zulassungsbehörde stellen. In Deutschland sind dies das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) in Bonn und das Paul-Ehrlich-Institut (PEI) in Langen bei Frankfurt am Main. Andere nationale Zulassungsbehörden können dann diese zunächst nur in einem Land gültige Zulassung der Medikamente nach einer kurzen Prüfung übernehmen.
Im Anschluss kann das Medikament in den Handel gelangen. Dieser Forschungs- und Entwicklungsprozess erfordert insgesamt viele Ressourcen und verursacht hohe Kosten. Unternehmen werden nur dann in die Entwicklung eines neuen Produktes zur Befriedigung eines unerfüllten medizinischen Bedarfs starten, wenn ihnen dies wirtschaftlich sinnvoll erscheint. Umgekehrt gibt es daher viele unerfüllte Bedarfe, für die derzeit keine neuen Arzneimittel entwickelt werden. Die europäischen und nationalen Gesetzgeber sind sich dessen bewusst und unterstützen die Entwicklung von Arzneimitteln beispielsweise für Kinder oder für Patienten mit seltenen Krankheiten mit Fördermitteln und Prämien.

Dr. med. Susanne Isfort,
Koordinierende Geschäftsführerin des CTC-A an der Uniklinik RWTH Aachen
Center for Translational & Clinical Research (CTC-A)
Das CTC-A wurde von der Medizinischen Fakultät der RWTH Aachen als operative Dienstleistungseinrichtung zur Unterstützung der patientenorientierten Forschung gegründet. Die Einrichtung wurde mit der Koordination klinischer Studien an der Uniklinik RWTH Aachen und der Entwicklung von IT-Strukturen für exzellente Qualität in klinischen Studien beauftragt. Das CTC-A arbeitet als zentrale Dachorganisation zur Steuerung und Integration industrieller und eigeninitiierter klinischer Forschung. Die Unterstützung der forschenden Einrichtungen durch das CTC-A erfolgt individuell und bedarfsgerecht. Die Aufgabenschwerpunkte liegen in der formalen und administrativen Unterstützung der Forscher bei Förderanträgen und Planung, Durchführung und Auswertung klinischer Studien sowie Vollkostenkalkulationen und Vertragsprüfungen und -verhandlungen. Die professionelle Unterstützung wird durch ein effizientes Schnittstellenmanagement gewährleistet. ![]()